Protein Sequence Classification
A case study on the Pfam dataset to classify protein families.

Abstract
Proteins are large, complex biomolecules that play many critical roles in biological bodies. Proteins are made up of one or more long chains of amino acid sequences. These Sequences are the arrangement of amino acids in a protein, held together by peptide bonds. Proteins can be made from 20 different kinds of amino acids, and the structure and function of each protein are determined by the kinds of amino acids used to make it and how they are arranged.
Understanding this relationship between amino acid sequence and protein function is a long-standing problem in molecular biology with far-reaching scientific implications. Can we use deep learning that learns the relationship between unaligned amino acid sequences and their functional annotations across all protein families of the Pfam database.
I created this project with Deepnote, an awesome product for data science professionals to run Jupyter notebooks over cloud. Super easy to setup, collaborative and offers many more features than Google Colab. Here is the complete notebook.
Problem Statement
- Classification of protein’s amino acid sequence to one of the protein family accession, based on Pfam dataset.
- In other words, the task is: given the amino acid sequence of the protein domain, predict which class it belongs to.
Data Overview
We have been provided with 5 features, they are as follows:
sequence: These are usually the input features to the model. Amino acid sequence for this domain. There are 20 very common amino acids (frequency > 1,000,000), and 4 amino acids that are quite uncommon: X, U, B, O, and Z.family_accession: These are usually the labels for the model. Accession number in form PFxxxxx.y (Pfam), where xxxxx is the family accession, and y is the version number. Some values of y are greater than ten, and so 'y' has two digits.sequence_name: Sequence name, in the form "uniprot_accession_id/start_index-end_index".aligned_sequence: Contains a single sequence from the multiple sequence alignment with the rest of the members of the family in seed, with gaps retained.family_id: One-word name for the family.
Source: Kaggle
Example Data point
sequence: HWLQMRDSMNTYNNMVNRCFATCIRSFQEKKVNAEEMDCTKRCVTKFVGYSQRVALRFAE
family_accession: PF02953.15
sequence_name: C5K6N5_PERM5/28-87
aligned_sequence: ....HWLQMRDSMNTYNNMVNRCFATCI...........RS.F....QEKKVNAEE.....MDCT....KRCVTKFVGYSQRVALRFAE
family_id: zf-Tim10_DDPMachine Learning Problem
It is a multi class classification problem, for a given sequence of amino acids we need to predict its protein family accession.
Metric
- Multi class log loss
- Accuracy
Exploratory Data Analysis
In this section, we will explore, visualize and try to understand the given features. Given data is already split into 3 folders i.e., train, dev, and test using random split.
First, let’s load the training, val, and test datasets.
The sizes of the given sets are as follows:
- Train size: 1086741 (80%)
- Val size: 126171 (10%)
- Test size: 126171 (10%)
NOTE: I have considered less data because of limited computational power. However, the same solution can also be scaled for the whole Pfam dataset.
Sequence Counts
First, let’s count the number of codes(amino acid) in each unaligned sequences.
- Most of the unaligned amino acid sequences have character counts in the range of 50–250.
Sequence Code Frequency
Let’s also find the frequency for each code(amino acid) in each unaligned sequences.
Plotting code frequency count for train, val, and test data

- The most frequent amino acid code is Leucine(L) followed by Alanine(A), Valine(V), and Glycine(G).
- As we can see, the uncommon amino acids (i.e., X, U, B, O, Z) are present in very less quantities. Therefore we can consider only 20 common natural amino acids for sequence encoding at the preprocessing step.
Text Preprocessing
Amino acid sequences are represented with their corresponding 1 letter code, for example, the code for alanine is (A), arginine is (R), and so on. The complete list of amino acids with their code can be found here.
Example, unaligned sequence:
PHPESRIRLSTRRDAHGMPIPRIESRLGPDAFARLRFMARTCRAILAAAGCAAPFEEFSSADAFSSTHVFGTCRMGHDPMRNVVDGWGRSHRWPNLFVADASLFPSSGGGESPGLTIQALALRT
For building deep learning models, we have to transform this textual data into a numerical form that the machines can process. I have used one hot encoding method for the same with considering 20 common amino acids as other uncommon amino acids are less in quantity.
The below code snippet creates a dictionary of considered 20 amino acids with integer values in incremental order to be further used for integer encoding.
For each unaligned amino acid sequences, 1 letter code is replaced by an integer value using the created code dictionary. If the code is not present in the dictionary the value is simply replaced by 0, thus considering only 20 common amino acids.
This step will convert 1 letter code sequence data into numerical data like this,
[13, 7, 13, 4, 16, 15, 8, 15, 10, 16, 17, 15, 15, 3, 1, 7, 6, 11, 13, 8, 13, 15, 8, 4, 16, 15, 10, 6, 13, 3, 1, 5, 1, 15, 10, 15, 5, 11, 1, 15, 17, 2, 15, 1, 8, 10, 1, 1, 1, 6, 2, 1, 1, 13, 5, 4, 4, 5, 16, 16, 1, 3, 1, 5, 16, 16, 17, 7, 18, 5, 6, 17, 2, 15, 11, 6, 7, 3, 13, 11, 15, 12, 18, 18, 3, 6, 19, 6, 15, 16, 7, 15, 19, 13, 12, 10, 5, 18, 1, 3, 1, 16, 10, 5, 13, 16, 16, 6, 6, 6, 4, 16, 13, 6, 10, 17, 8, 14, 1, 10, 1, 10, 15, 17]
Next post padding is done with a max sequence length of 100 which pads with 0 if the total sequence length is less than 100 else truncates the sequence up to a max length of 100.
Finally, each code in the sequences is converted into a one hot encoded vector.
Deep Learning Models
I have referred to this paper for defining model architectures and trained two separate models, one is bidirectional LSTM and the other one is inspired by ResNet, a CNN based architecture.
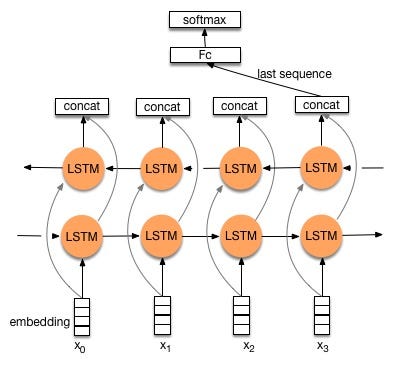
Model 1: Bidirectional LSTM
Variations of Recurrent neural networks like LSTMs are the first choice when working on NLP based problems as they were made to work with temporal or sequential data like text.
RNNs are a type of Neural Network where the outputs from previous steps are fed as inputs to the current step, thus remembering some information about the sequence. RNNs are great when it comes to short contexts, but it has a limitation in remembering longer sequences because of the vanishing gradient problem.
LSTM (Long Short-Term Memory) Networks are improved versions of RNN, specialized in remembering information for an extended period using a gating mechanism which makes them selective in what previous information to be remembered, what to forget and how much current input is to be added for building the current cell state.

Unidirectional LSTM only preserves the information from the past because the inputs it has seen are from the past. Using bidirectional will run the inputs in two ways, one from past to future and one from future to past allowing it to preserve contextual information from both past and future at any point of time.
A more in-depth explanation for RNN and LSTM can be found here.
Model architecture follows like this, first there is an embedding layer that learns the vector representation for each code followed by bidirectional LSTM. For regularization, Dropout is added to prevent model over-fitting.
The output layer i.e., the softmax layer will give probability values for all the unique classes(1000), and based on the highest predicted probability, the model will classify amino acid sequences to one of its protein family accession.
The model is trained with 33 epochs, batch_size of 256, and was able to achieve a loss of (0.386) with (95.8%) accuracy for test data.
439493/439493 [==============================] - 28s 65us/step
Train loss: 0.36330516427409587
Train accuracy: 0.9645910173696531
----------------------------------------------------------------------
54378/54378 [==============================] - 3s 63us/step
Val loss: 0.3869630661736021
Val accuracy: 0.9577034830108782
----------------------------------------------------------------------
54378/54378 [==============================] - 3s 64us/step
Test loss: 0.3869193921893196
Test accuracy: 0.9587149214887501Model 2: ProtCNN
This model uses residual blocks inspired by ResNet architecture which also includes dilated convolutions offering a larger receptive field without increasing the number of model parameters.
ResNet (Residual Networks)
Deeper neural networks are difficult to train because of the vanishing gradient problem — as the gradient is back-propagated to earlier layers, repeated multiplication may make the gradient infinitely small. As a result, as the network goes deeper, its performance gets saturated or even starts degrading rapidly.

ResNets introduces skip connection or identity shortcut connection which is adding initial input to the output of the convolution block.

This mitigates the problem of vanishing gradient by allowing the alternate shortcut path for the gradient to flow through. It also allows the model to learn an identity function which ensures that the higher layer will perform at least as well as the lower layer, and not worse.
Dilated Convolution

Dilated convolutions introduce another parameter to convolutional layers called dilation rate. This defines the spacing between the values in a kernel. A 3x3 kernel with a dilation rate of 2 will have the same field of view as a 5x5 kernel, while only using 9 parameters. Imagine taking a 5x5 kernel and deleting every second column and row.
Dilated convolutions deliver a wider field of view at the same computational cost.
ProtCNN Model Architecture

Amino acids sequences are converted to one hot encoding with shape (batch_size, 100, 21) as input to the model. The initial convolution operation is applied to the input with a kernel size of 1 to extract basic properties. Then two identical residual blocks are used to capture complex patterns in the data which are inspired by the ResNet architecture, this will help us to train the model with more number of epochs and with better model performance.
I have defined residual block slight differently from the ResNet paper. Instead of performing three convolutions only two convolutions are used with also adding one more parameter (dilation rate) to the first convolution so as to have a wider field of view with the same number of model parameters.
Each Convolution operation in the residual block follows a basic pattern of BatchNormalization => ReLU => Conv1D. In the residual block, the first convolution is done with a kernel size of 1x1 with a dilation rate, and the second convolution operation is done with a larger kernel size of 3x3.
Finally, after applying the convolution operations, a skip connection is formed by adding the initial input(shortcut) and the output from the applied convolution operations.
After two residual blocks, Max pooling is applied for reducing the spatial size of the representation. For regularization, Dropout is added to prevent model over-fitting.
This model is trained with 10 epochs, batch_size of 256, and validated on the validation data.

439493/439493 [==============================] - 38s 85us/step
Train loss: 0.3558084576734698
Train accuracy: 0.9969123512774948
----------------------------------------------------------------------
54378/54378 [==============================] - 5s 85us/step
Val loss: 0.39615299251274316
Val accuracy: 0.9886718893955224
----------------------------------------------------------------------
54378/54378 [==============================] - 5s 85us/step
Test loss: 0.3949931418234982
Test accuracy: 0.9882489242257847As we can see, the results of this model are better than the Bidirectional LSTM model. More improvements to this can be achieved by taking a majority vote across an ensemble of ProtCNN models.
Conclusion
In this case study, we have explored deep learning models that learn the relationship between unaligned amino acid sequences and their functional annotations. The ProtCNN model has achieved significant results which are more accurate and computationally efficient than the current state-of-the-art techniques like BLASTp to annotate protein sequences.
These results suggest deep learning models will be a core component of future protein function prediction tools.
Thank you for reading. The complete code can be found here.

{kind=link}